By Shari M. Ling, M.D. and Joan M. Bathon, M.D.
- OA is primarily a disease of cartilage
- Is OA simply a process of aging of cartilage?
- What molecules are responsible for degrading cartilage matrix?
- What factor(s) is responsible for inducing metalloprotease synthesis?
- Can cartilage repair itself?
- Summary
- References
Although epidemiological studies have promoted our understanding of the risk factors that predispose to OA, we do not yet understand the initiating events that trigger the disease.
OA is primarily a disease of cartilage
Cartilage is a unique tissue with viscoelastic and compressive properties which are imparted by its extracellular matrix, composed predominantly of type II collagen and proteoglycans.

Under normal conditions, this matrix is subjected to a dynamic remodeling process in which low levels of degradative and synthetic enzyme activities are balanced, such that the volume of cartilage is maintained. In OA cartilage, however, matrix degrading enzymes are overexpressed, shifting this balance in favor of net degradation, with resultant loss of collagen and proteoglycans from the matrix.
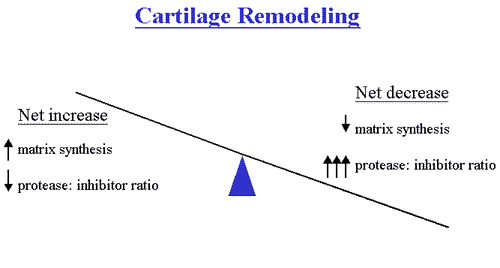
Presumably in response to this loss, chondrocytes initially proliferate and synthesize enhanced amounts of proteoglycan and collagen molecules. As the disease progresses, however, reparative attempts are outmatched by progressive cartilage degradation. Fibrillation, erosion and cracking initially appear in the superficial layer of cartilage and progress over time to deeper layers, resulting eventually in large clinically observable erosions. OA, in simplistic terms, therefore, can be thought of as a process of progressive cartilage matrix degradation to which an ineffectual attempt at repair is made.
Is OA simply a process of aging of cartilage?
A critical question is whether OA is truly a disease or a natural consequence of aging. Several differences between aging cartilage and OA cartilage have been described, suggesting the former. For example, although denatured type II collagen is found in both normal aging and OA cartilage, it is more predominant in OA. In addition, OA and normal aging cartilage differ in the amount of water content and the in ratio of chondroitin-sulfate to keratin sulfate constituents. The expression of a chondroitin-sulfate epitope (epitope 846) in OA cartilage, that is otherwise only present in fetal and neonatal cartilage, provides further evidence that OA is a distinct pathologic process. A final but important distinction is that degradative enzyme activity is increased in OA, but not in normal aging cartilage.
What molecules are responsible for degrading cartilage matrix?
The primary enzymes responsible for the degradation of cartilage are the matrix metalloproteinases (MMPs).

These enzymes are secreted by both synovial cells and chondrocytes and are categorized into three general categories: a) collagenases; b) stromelysins; and, c) gelatinases. Under normal conditions, MMP synthesis and activation are tightly regulated at several levels. They are secreted as inactive proenzymes that require enzymatic cleavage in order to become activated. Once activated, MMPs become susceptible to the plasma-derived MMP inhibitor, alpha-2-macroglobulin, and to tissue inhibitors of MMPs (TIMPs) that are also secreted by synovial cells and chondrocytes. In OA, synthesis of MMPs is greatly enhanced and the available inhibitors are overwhelmed, resulting in net degradation. Interestingly, stromelysin can serve as an activator for its own proenzyme, as well as for procollagenase and prostromelysin, thus creating a positive feedback loop of proMMP activation in cartilage.
What factor(s) is responsible for inducing metalloprotease synthesis?
One candidate is interleukin-1 (IL-1). IL-1 is a potent pro-inflammatory cytokine that, in vitro, is capable of inducing chondrocytes and synovial cells to synthesize MMPs.

Furthermore, IL-1 suppresses the synthesis of type II collagen and proteoglycans, and inhibits transforming growth factor-ß stimulated chondrocyte proliferation. The presence of IL-1 RNA and protein have been confirmed in OA joints. Thus, IL-1 may not only actively promote cartilage degradation, but may also suppress attempts at repair, in OA. In addition to these effects, IL-1 induces nitric oxide production, chondrocyte apoptosis, and prostaglandin synthesis, which further contribute to cartilage deterioration. Under normal conditions, an endogenous IL-1 receptor antagonist regulates IL-1 activity. A relative excess of IL-1 and/or deficiency of the IL-1 receptor antagonist could conceivably result in the cartilage destruction that is characteristic of OA. It is likely that other cytokines or particulate material from damaged cartilage may also contribute to this inflammatory, degradative process.
Can cartilage repair itself?
Growth factors are produced locally in cartilage and synovium and are likely to contribute to local cartilage remodeling by stimulating the de novo synthesis of collagen and proteoglycans. Transforming growth factor ß (TGFß) is the best characterized and most potent of the chondrocyte growth factors. Not only does TGFß stimulate de novo matrix synthesis, but it also counteracts cartilage degradation by down regulating IL-1 receptor expression and by increasing IL-1 receptor antagonist release and TIMP expression. Insulin-like growth factor (IGF-1) and basic fibroblast growth factor (b-FGF) are also present in OA cartilage and likely to contribute to reparative attempts, although, as noted, degradation ultimately outstrips repair in OA cartilage.
Summary
In summary, MMPs and pro-inflammatory cytokines (e.g., IL-1) appear to be important mediators of cartilage destruction in OA. Synthesis and secretion of growth factors and of inhibitors of MMPs and cytokines are apparently inadequate to counteract these degradative forces. Progressive cartilage degradation and OA result. New therapies (see Treatment), focused on reducing MMP activity and on stimulating matrix synthesis, are in development.
References
- Lohmander LS et al. Cartilage matrix metabolism in osteoarthritis: markers in synovial fluid, serum, and urine. Clin Biochem 25:167-1674,1992.
- Hollander AP et al. Increased damage to type II collagen in osteoarthritic articular cartilage detected by a new immunoassay. J Clin Invest 93:1722-1732, 1994.
- Poole AR et al. Changes in cartilage metabolism in arthritis are reflected by altered serum and synovial fluid levels of the cartilage proteoglycan aggrecan, implications for pathogenesis. J Clin Invest 94:25-33, 1994
- Woessner Jr. MF, Gunja-Smith Z., Role of metalloproteinases in human osteoarthritis. J Rheumatol 18;99-101, 1991
- Lotz M et al. Cytokine regulation of chondrocyte functions. J Rheumatol 22:104-108, 1995.
- Guerne P-A, et al. Growth factor responsiveness of human articular chondrocytes in aging and development. Arthritis Rheum 38:960-968, 1995
- Sharif M et al. Serum hyaluronic acid level as a predictor of disease progression in osteoarthritis of the knee. Arthritis Rheum 38:760-767, 1995
- Hochberg MC et al. Serum levels of insulin-like growth factor 1 in subjects with osteoarthritis of the knee. Arthritis Rheum 37:1177-1180, 1994
- Campion GP, Delmas P, Dieppe P. Serum and synovial fluid osteocalcin (bone gla protein) levels in joint diseases. Br J Rheumatol 28:393-398, 1989
- Caterson B. Immunological aspects of markers of joint disease. J Rheumatol 18:19-26, 1991.
